

TOMBO is a unique first-principles program using the all-electron mixed basis approach. So, it accurately describes both localized core-electron states and extended free-electron-like states. Moreover, it can treat not only periodic systems (supercell systems and crystals) but also isolated systems (atoms, clusters, and molecules). Local density approximation (LDA) of density functional theory (DFT) is default. But, Hartree-Fock (HF) approximation is possible too. In addition, GW, Bethe-Salpeter equation (BSE), TDGW, T-matrix, etc. are possible. So, TOMBO is a very powerful program, in particular, for all-electron excited state calculations.
All-electron mixed basis approach
The all-electron mixed basis approach uses both plane waves (PWs) and atomic orbitals (AOs) as a basis set1. Therefore, it can well describe both extended states and localized states. Moreover, TOMBO has no basis set superposition error (BSSE) and reduces the overcompleteness problem. This is because AOs are confined inside non-overlapping atomic spheres1. Therefore one can avoid unnecessary numerical error caused by the computation of overlap integrals between adjacent AOs.
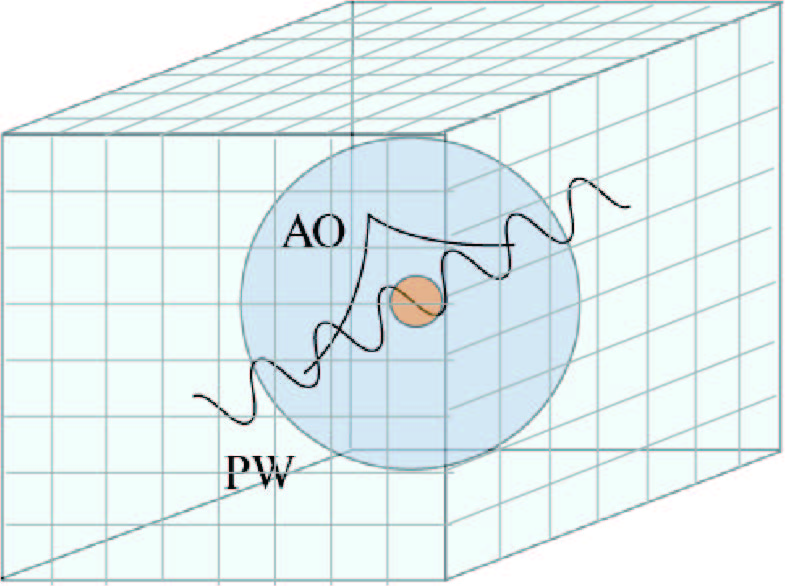
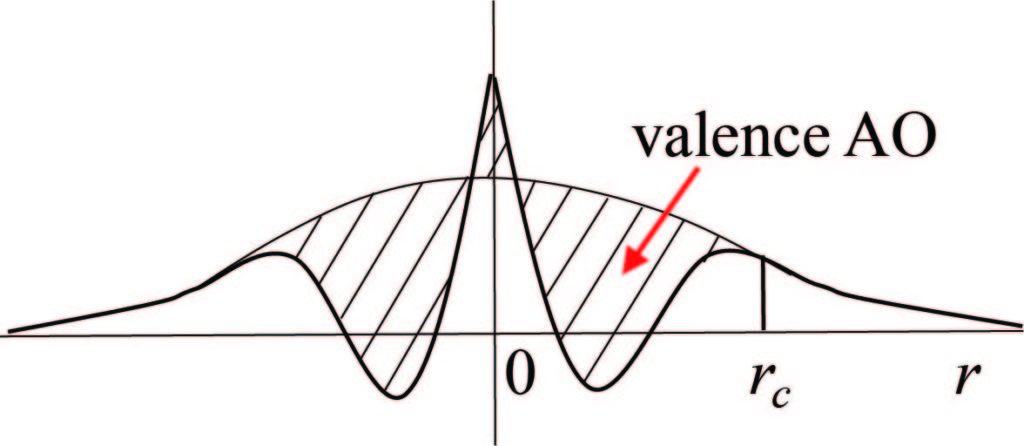

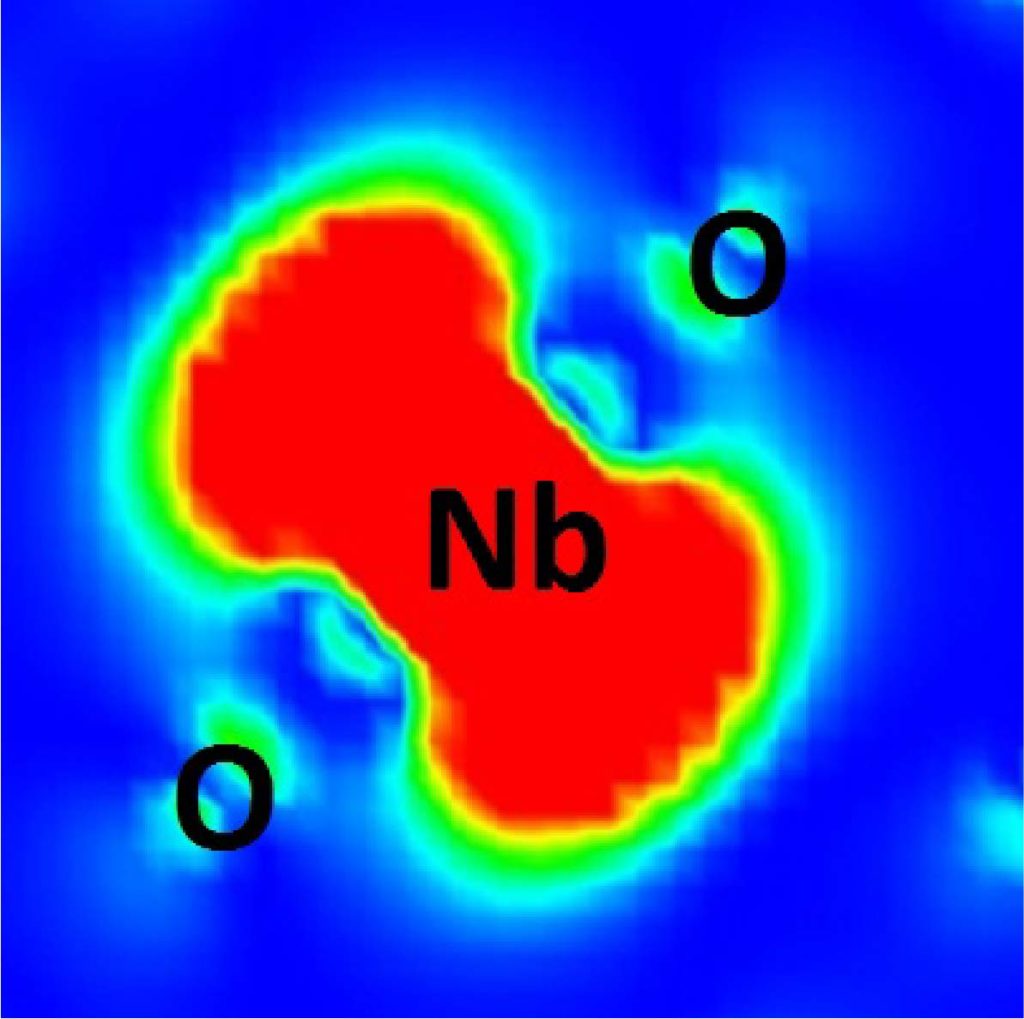
[1] S. Ono, Y. Noguchi, R. Sahara, Y. Kawazoe, and K. Ohno, Computer Physics Communications 189, 20 (2015).
[2] M. Zhang, S. Ono, and K. Ohno, Physical Review B 92, 035205 (2015). — GW for Nb impurity in TiO2
Excited State Calculations
Furthermore, TOMBO can perfectly perform excited state calculations3-6,8. This is the most important characteristic of TOMBO. We can do it because of extended quasiparticle (EQP) theory4,8. Extended Kohn-Sham (EKS) theory7,8, recently established in our group, guarantees the calculations. Therefore, TDGW molecular dynamics simulation3, GW photoabsorption calculation for cations without solving BSE5, and GW + BSE calculation with a core hole for X-ray emission spectra (XES)6,8 and resonant inelastic X-ray scattering (RIXS)8 are possible in TOMBO.
See also ・Excited State Algorithm
[3] T. N. Pham, S. Ono, and K. Ohno, Journal of Chemical Physics 144, 144309 (2016). — TDGW
[4] K. Ohno, S. Ono, and T. Isobe, Journal of Chemical Physics 146, 084108 (2017). — EQP theory
[5] T. Isobe, R. Kuwahara, and K. Ohno, Physical Review A 97, 060502(R) (2018). — GW for Cations
[6] T. Aoki and K. Ohno, Physical Review B 100, 075149 (2019). — XES by GW + BSE with a core hole
[7] T. Nakashima, H. Raebiger, and K. Ohno, Physical Review B 104, L201116 (2021). — EKS theory
[8] K. Ohno and T. Aoki, Physical Chemistry Chemical Physics 24, 16586-16595 (2022). — EQP/EKS theories, XES & RIXS